The Department of Anesthesiology at the University of Illinois College of Medicine was established by Dr. Mac Samual Sadove over 70 years ago. Our institution has a legacy of excellent patient care, research, and clinical training in anesthesiology. We are a leading clinical and research Anesthesiology department, offering a broad range of clinical services and research opportunities. Much of our success stems from the experience and diversity of our faculty. Many of our graduates are innovators and leaders within our specialty.
We're a highly sought after department Heading link
-
Top 20 for NIH funding, nationally.
-
7 Labs
-
4 Fellowships
Education Heading link
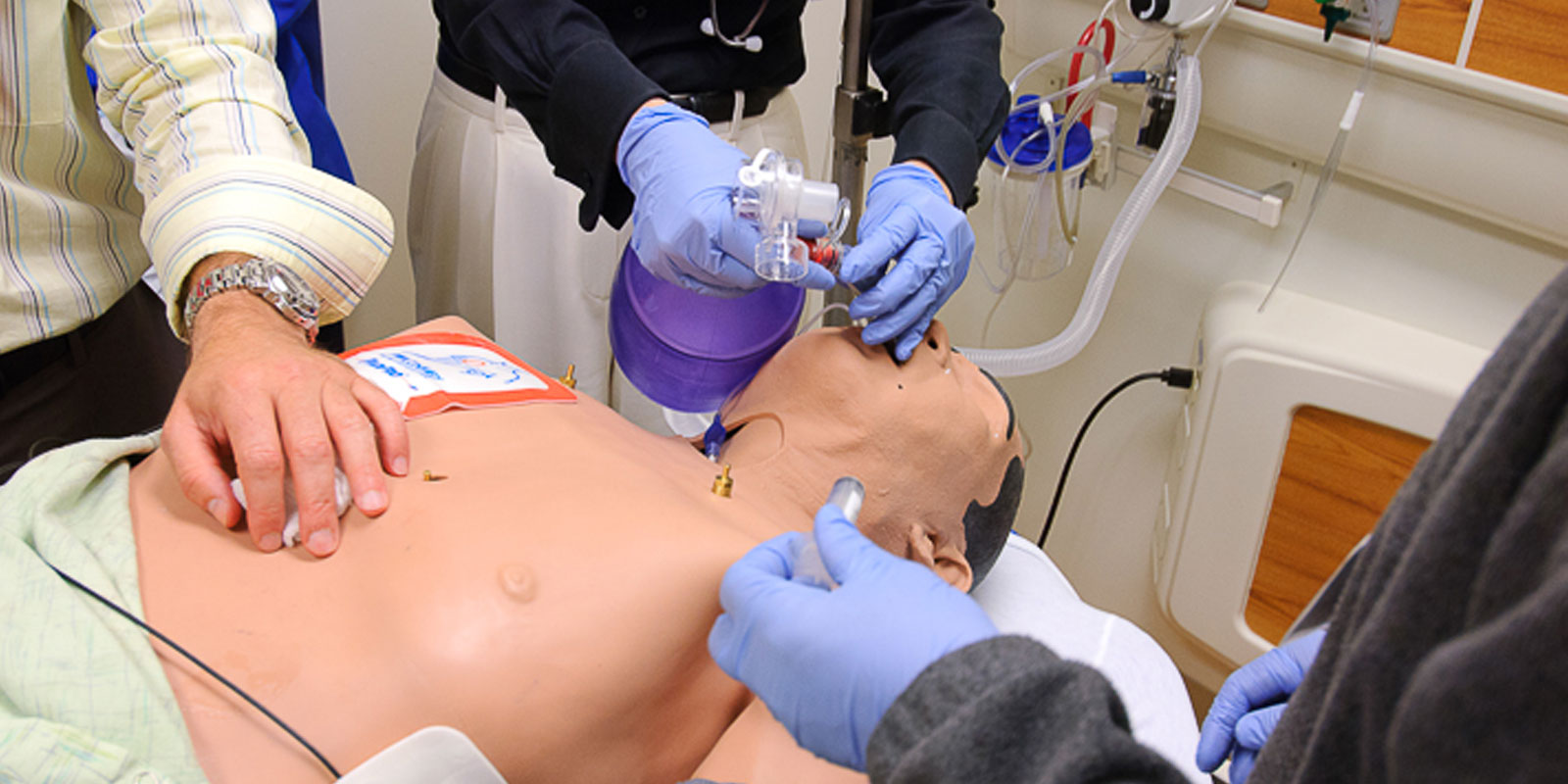
Learn more about our residency program
Helpful residency links
Learn about the four fellowships in the Department of Anesthesiology
Helpful fellowship links
Offerings for medical students
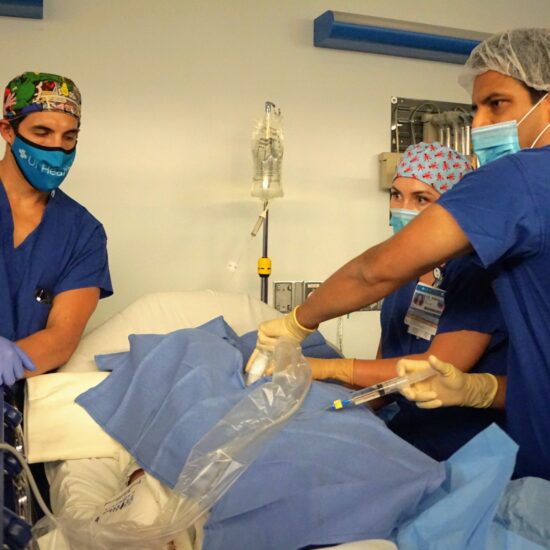
Research Heading link
Distinguished as the top-ranked Anesthesiology department in Illinois for total NIH grant funding. Our research endeavors are further supported by various foundations, solidifying our commitment to advancing knowledge and innovation in the field.
Department News Heading link

