Our Programs Heading link

Start your career in Pharmacy
Bachelor of Science in Pharmaceutical Sciences
Flexible, online options for healthcare professionals
Online Programs
Become a Pharmacist at UIC
Leading the field in research
Earn a PhD in Pharmacy or Pharmaceutical Sciences
Apply to be a resident or fellow at UIC
Residency and Fellowships
Points of Pride Heading link
-
#1 School of Pharmacy in Illinois, according to US News & World Report
-
# 7 total research grant funding according to the American Association of Colleges of Pharmacy (AACP)
-
1859 Year the college was founded, one of the oldest pharmacy schools in the country

Become a Pharmacist Heading link
News Heading link


Events Heading link
MIKIW 2024 Conference
UIC Spring Commencement | College of Pharmacy
UIC Pharmacy Rockford Campus celebrates 10 years of graduates
White Coat Ceremony | Class of 2028
One College, Two Campuses, Unlimited Opportunities Heading link


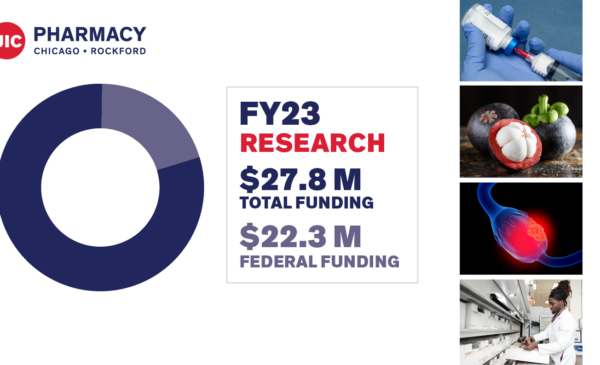
By The Numbers Heading link
When you choose UIC College of Pharmacy, you choose a great education, world-class faculty, cutting-edge research, and a commitment to service.
Visit our “By The Numbers” page to get an overview on what we offer, including facts and figures that all add up to one thing: an amazing education at a top institution.

Diversity, Equity and Inclusion Heading link
We strive to be a community where differences are embraced, that emphasizes inclusion and recognizes diversity as a strength, and where everyone works together to be the global leader in innovative pharmacy education, research, and practice to improve human health.